There’s a set of videos that walks through each section below. To make it easier for you to jump around the video examples, I cut the long video into smaller pieces and included them all in one YouTube playlist.
You can also watch the playlist (and skip around to different sections) here:
Program background
Let’s revisit the mosquito net program we used in the DAG example. Here, researchers are interested in whether using mosquito nets decreases an individual’s risk of contracting malaria. They have collected data from 1,752 households in an unnamed country and have variables related to environmental factors, individual health, and household characteristics. The data is not experimental—researchers have no control over who uses mosquito nets, and individual households make their own choices over whether to apply for free nets or buy their own nets, as well as whether they use the nets if they have them.
The CSV file contains the following columns:
Malaria risk (malaria_risk): The likelihood that someone in the household will be infected with malaria. Measured on a scale of 0–100, with higher values indicating higher risk.
Mosquito net (net and net_num): A binary variable indicating if the household used mosquito nets.
Eligible for program (eligible): A binary variable indicating if the household is eligible for the free net program.
Income (income): The household’s monthly income, in US dollars.
Nighttime temperatures (temperature): The average temperature at night, in Celsius.
Health (health): Self-reported healthiness in the household. Measured on a scale of 0–100, with higher values indicating better health.
Number in household (household): Number of people living in the household.
Insecticide resistance (resistance): Some strains of mosquitoes are more resistant to insecticide and thus pose a higher risk of infecting people with malaria. This is measured on a scale of 0–100, with higher values indicating higher resistance.
Our goal
Our goal in this example is to estimate the causal effect of bed net usage on malaria risk using only observational data. This was not an RCT, so it might seem a little sketchy to make claims of causality. But if we can draw a correct DAG and adjust for the correct nodes, we can isolate the net → malaria relationship and talk about causality.
Because this is simulated data, we know the true causal effect of the net program because I built it into the data. The true average treatment effect (ATE) is -10. Using a mosquito net causes the risk of malaria to decrease by 10 points, on average.
Let’s see if we can find that 10 point effect!
Load data and libraries
First, let’s download the dataset (if you haven’t already), put in a folder named data, and load it:
library(tidyverse) # ggplot(), %>%, mutate(), and friendslibrary(ggdag) # Make DAGslibrary(dagitty) # Do DAG logic with Rlibrary(broom) # Convert models to data frameslibrary(MatchIt) # Match thingslibrary(modelsummary) # Make side-by-side regression tablesset.seed(1234) # Make all random draws reproducible
Code
# Load the data.# It'd be a good idea to click on the "nets" object in the Environment panel in# RStudio to see what the data looks like after you load itnets <-read_csv("data/mosquito_nets.csv")
DAG and adjustment sets
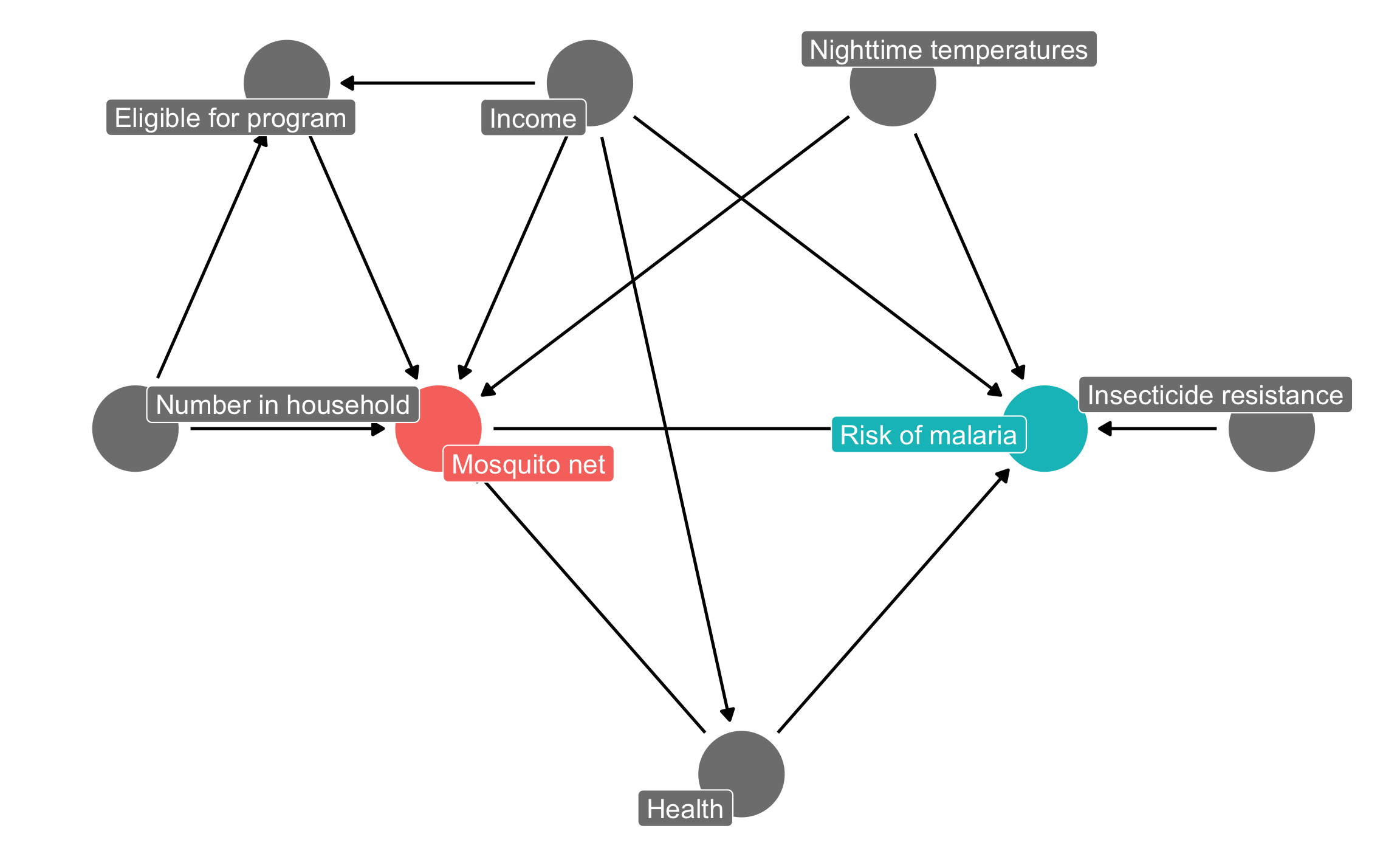
Before running any models, we need to find what we need to adjust for. Recall from the DAG example that we built this causal model to show the data generating process behind the relationship between mosquito net usage and malaria risk:
Code
mosquito_dag <-dagify( malaria_risk ~ net + income + health + temperature + resistance, net ~ income + health + temperature + eligible + household, eligible ~ income + household, health ~ income,exposure ="net",outcome ="malaria_risk",coords =list(x =c(malaria_risk =7, net =3, income =4, health =5,temperature =6, resistance =8.5, eligible =2, household =1),y =c(malaria_risk =2, net =2, income =3, health =1,temperature =3, resistance =2, eligible =3, household =2)),labels =c(malaria_risk ="Risk of malaria", net ="Mosquito net", income ="Income",health ="Health", temperature ="Nighttime temperatures",resistance ="Insecticide resistance",eligible ="Eligible for program", household ="Number in household"))ggdag_status(mosquito_dag, use_labels ="label", text =FALSE) +guides(fill ="none", color ="none") +# Disable the legendtheme_dag()
Following the logic of do-calculus, we can find all the nodes that confound the relationship between net usage and malaria risk, since those nodes open up backdoor paths and distort the causal effect we care about. We can either do this graphically by looking for any node that points to both net and malaria risk, or we can use R:
Code
adjustmentSets(mosquito_dag)## { health, income, temperature }
Based on the relationships between all the nodes in the DAG, adjusting for health, income, and temperature is enough to close all backdoors and identify the relationship between net use and malaria risk.
Naive correlation-isn’t-causation estimate
For fun, we can calculate the difference in average malaria risk for those who did/didn’t use mosquito nets. This is most definitely not the actual causal effect—this is the “correlation is not causation” effect that doesn’t account for any of the backdoors in the DAG.
We can do this with a table (but then we have to do manual math to subtract the FALSE average from the TRUE average):
Code
nets %>%group_by(net) %>%summarize(number =n(),avg =mean(malaria_risk))## # A tibble: 2 × 3## net number avg## <lgl> <int> <dbl>## 1 FALSE 1071 41.9## 2 TRUE 681 25.6
According to this estimate, using a mosquito net is associated with a 16.33 point decrease in malaria risk, on average. We can’t legally talk about this as a causal effect though—there are confounding variables to deal with.
Matching
We can use matching techniques to pair up similar observations and make the unconfoundedness assumption—that if we see two observations that are pretty much identical, and one used a net and one didn’t, the choice to use a net was random.
Because we know from the DAG that income, nighttime temperatures, and health help cause both net use and malaria risk (and confound that relationship!), we’ll try to find observations with similar values of income, temperatures, and health that both used and didn’t use nets.
We can use the matchit() function from the MatchIt R package to match points based on Mahalanobis distance. There are lots of other options available—see the online documentation for details.
We can include the replace = TRUE option to make it so that points that have been matched already can be matched again (that is, we’re not forcing a one-to-one matching; we have one-to-many matching instead).
Step 1: Preprocess
Code
matched_data <-matchit(net ~ income + temperature + health,data = nets,method ="nearest",distance ="mahalanobis",replace =TRUE)summary(matched_data)## ## Call:## matchit(formula = net ~ income + temperature + health, data = nets, ## method = "nearest", distance = "mahalanobis", replace = TRUE)## ## Summary of Balance for All Data:## Means Treated Means Control Std. Mean Diff. Var. Ratio eCDF Mean eCDF Max## income 955 873 0.41 1.4 0.104 0.198## temperature 23 24 -0.17 1.1 0.042 0.097## health 55 48 0.36 1.2 0.071 0.168## ## Summary of Balance for Matched Data:## Means Treated Means Control Std. Mean Diff. Var. Ratio eCDF Mean eCDF Max Std. Pair Dist.## income 955 950 0.025 1.1 0.009 0.048 0.12## temperature 23 23 -0.006 1.0 0.007 0.026 0.12## health 55 55 0.019 1.1 0.007 0.026 0.12## ## Sample Sizes:## Control Treated## All 1071 681## Matched (ESS) 321 681## Matched 436 681## Unmatched 635 0## Discarded 0 0
Here we can see that all 681 of the net users were paired with similar-looking non-users (439 of them). 632 people weren’t matched and will get discarded. If you’re curious, you can see which treated rows got matched to which control rows by running matched_data$match.matrix.
We can create a new data frame of those matches with match.data():
Code
matched_data_for_real <-match.data(matched_data)
Step 2: Estimation
Now that the data has been matched, it should work better for modeling. Also, because we used income, temperatures, and health in the matching process, we’ve adjusted for those DAG nodes and have closed those backdoors, so our model can be pretty simple here:
The 12.73 point decrease here is better than the naive estimate, but it’s not the true 10 point causal effect (that I built in to the data). Perhaps that’s because the matches aren’t great, or maybe we threw away too much data. There are a host of diagnostics you can look at to see how well things are matched (check the documentation for MatchIt for examples.)
Actually, the most likely culprit for the incorrect estimate is that there’s some imbalance in the data. Because we set replace = TRUE, we did not do 1:1 matching—untreated observations were paired with more than one treated observation. As a result, the multiply-matched observations are getting overcounted and have too much importance in the model. Fortunately, matchit() provides us with a column called weights that allows us to scale down the overmatched observations when running the model. Importantly, these weights have nothing to do with causal inference or backdoors or inverse probability weighting—their only purpose is to help scale down the imbalance arising from overmatching. If you use replace = FALSE and enforce 1:1 matching, the whole weights column will just be 1.
We can incorporate those weights into the model and get a more accurate estimate:
After weighting to account for under- and over-matching, we find a -10.48 point causal effect. That’s much better than any of the other estimates we’ve tried so far! The reason it’s accurate is because we’ve closed the confounding backdoors and isolated the arrow between net use and malaria risk.
Inverse probability weighting
One potential downside to matching is that you generally have to throw away a sizable chunk of your data—anything that’s unmatched doesn’t get included in the final matched data.
An alternative approach to matching is to assign every observation some probability of receiving treatment, and then weight each observation by its inverse probability—observations that are predicted to get treatment and then don’t, or observations that are predicted to not get treatment and then do will receive more weight than the observations that get/don’t get treatment as predicted.
Generating these inverse probability weights requires a two step process: (1) we first generate propensity scores, or the probability of receiving treatment, and then (2) we use a special formula to convert those propensity scores into weights. Once we have inverse probability weights weights, we can incorporate them into our regression model.
Oversimplified crash course in logistic regression
There are many ways to generate propensity scores (like logistic regression, probit regression, and even machine learning techniques like random forests and neural networks), but logistic regression is probably the most common method.
The complete technical details of logistic regression are beyond the scope of this class, but if you’re curious you should check out this highly accessible tutorial.
All you really need to know is that the outcome variable in logistic regression models must be binary, and the explanatory variables you include in the model help explain the variation in the likelihood of your binary outcome. The Y (or outcome) in logistic regression is a logged ratio of probabilities, which forces the model’s output to be in a 0-1 range:
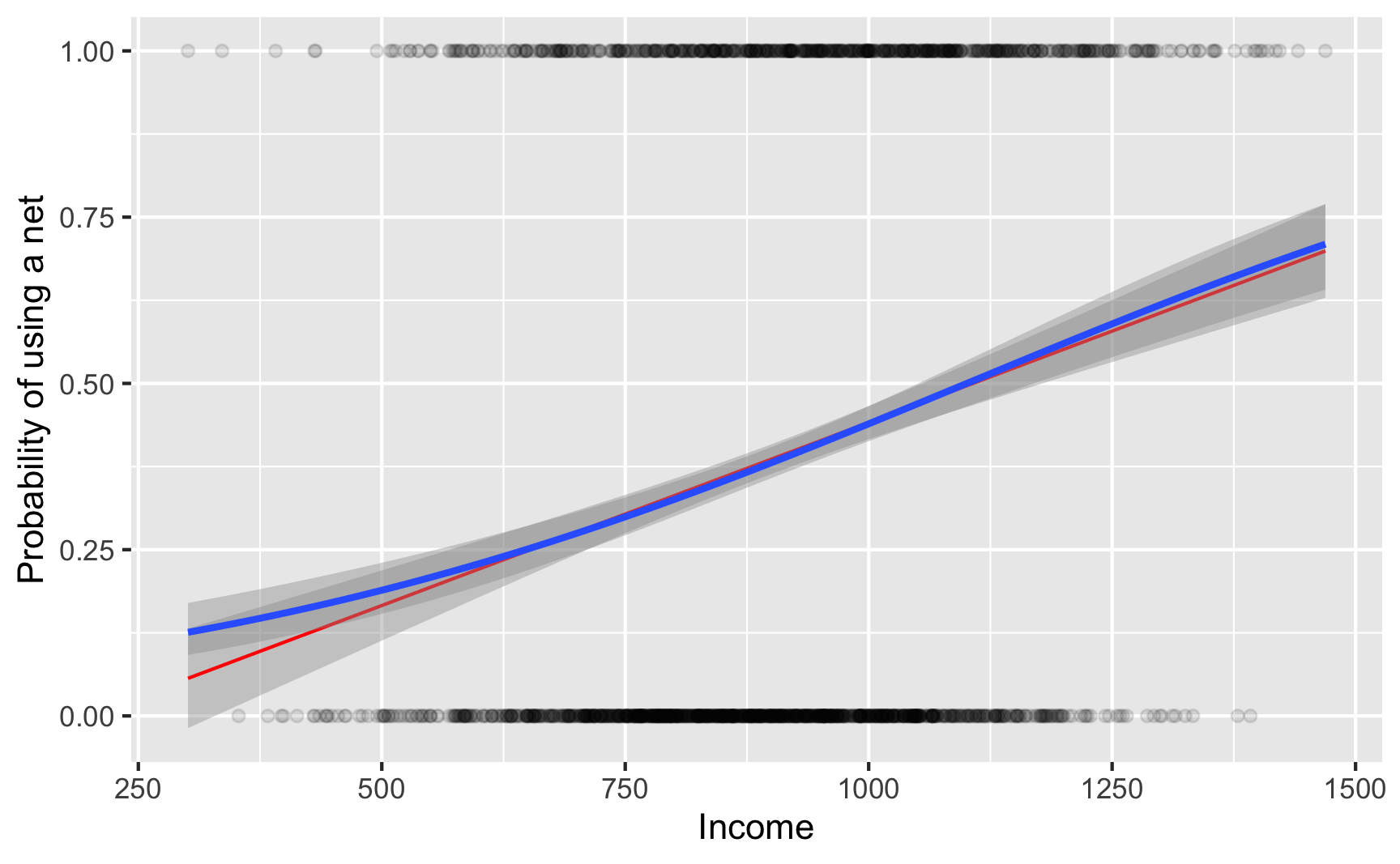
Here’s what it looks like visually. Because net usage is a binary outcome, there are lines of observations at 0 and 1 along the y axis. The blue curvy line here shows the output of a logistic regression model—people with low income have a low likelihood of using a net, while those with high income are far more likely to do so.
I also included a red line showing the results from a regular old lm() OLS model. It follows the blue line fairly well for a while, but predicts negative probabilities if you use lower values of income, like less than 250. For strange historical and mathy reasons, many economists like using OLS on binary outcomes (they even have a fancy name for it: linear probability models (LPMs)), but I’m partial to logistic regression since it doesn’t generate probabilities greater than 100% or less than 0%. (BUT DON’T EVER COMPLAIN ABOUT LPMs ONLINE. You’ll start battles between economists and other social scientists. 🤣)
Code
ggplot(nets, aes(x = income, y = net_num)) +geom_point(alpha =0.1) +geom_smooth(method ="lm", color ="red", size =0.5) +geom_smooth(method ="glm", method.args =list(family =binomial(link ="logit"))) +labs(x ="Income", y ="Probability of using a net")
The coefficients from a logistic regression model are interpreted differently than you’re used to (and their interpretations can be controversial!). Here’s an example for the model in the graph above:
Code
# Notice how we use glm() instead of lm(). The "g" stands for "generalized"# linear model. We have to specify a family in any glm() model. You can# technically run a regular OLS model (like you do with lm()) if you use# glm(y ~ x1 + x2, family = gaussian(link = "identity")), but people rarely do that.## To use logistic regression, you have to specify a binomial/logit family like so:# family = binomial(link = "logit")model_logit <-glm(net ~ income + temperature + health,data = nets,family =binomial(link ="logit"))tidy(model_logit)## # A tibble: 4 × 5## term estimate std.error statistic p.value## <chr> <dbl> <dbl> <dbl> <dbl>## 1 (Intercept) -1.32 0.376 -3.50 0.000464 ## 2 income 0.00209 0.000421 4.95 0.000000727## 3 temperature -0.0589 0.0125 -4.70 0.00000264 ## 4 health 0.00688 0.00430 1.60 0.109
The coefficients here aren’t normal numbers—they’re called “log odds” and represent the change in the logged odds as you move explanatory variables up. For instance, here the logged odds of using a net increase by 0.00688 for every one point increase in your health score. But what do logged odds even mean?! Nobody knows.
You can make these coefficients slightly more interpretable by unlogging them and creating something called an “odds ratio.” These coefficients were logged with a natural log, so you unlog them by raising \(e\) to the power of the coefficient. The odds ratio for temperature is \(e^{-0.0589}\), or 0.94. Odds ratios get interpreted a little differently than regular model coefficients. Odds ratios are all centered around 1—values above 1 mean that there’s an increase in the likelihood of the outcome, while values below 1 mean that there’s a decrease in the likelihood of the outcome.
Our nighttime temperature odds ratio here is 0.94, which is 0.06 below 1, which means we can say that for every one point increase in nighttime temperatures, a person is 6% less likely to use a net. If the coefficient was something like 1.34, we could say that they’d be 34% more likely to use a net; if it was something like 5.02 we could say that they’d be 5 times more likely to use a net; if it was something like 0.1, we could say that they’re 90% less likely to use a net.
You can make R exponentiate the coefficients automatically by including exponentiate = TRUE in tidy():
Code
tidy(model_logit, exponentiate =TRUE)## # A tibble: 4 × 5## term estimate std.error statistic p.value## <chr> <dbl> <dbl> <dbl> <dbl>## 1 (Intercept) 0.268 0.376 -3.50 0.000464 ## 2 income 1.00 0.000421 4.95 0.000000727## 3 temperature 0.943 0.0125 -4.70 0.00000264 ## 4 health 1.01 0.00430 1.60 0.109
BUT AGAIN this goes beyond the scope of this class! Just know that when you build a logistic regression model, you’re using explanatory variables to predict the probability of an outcome.
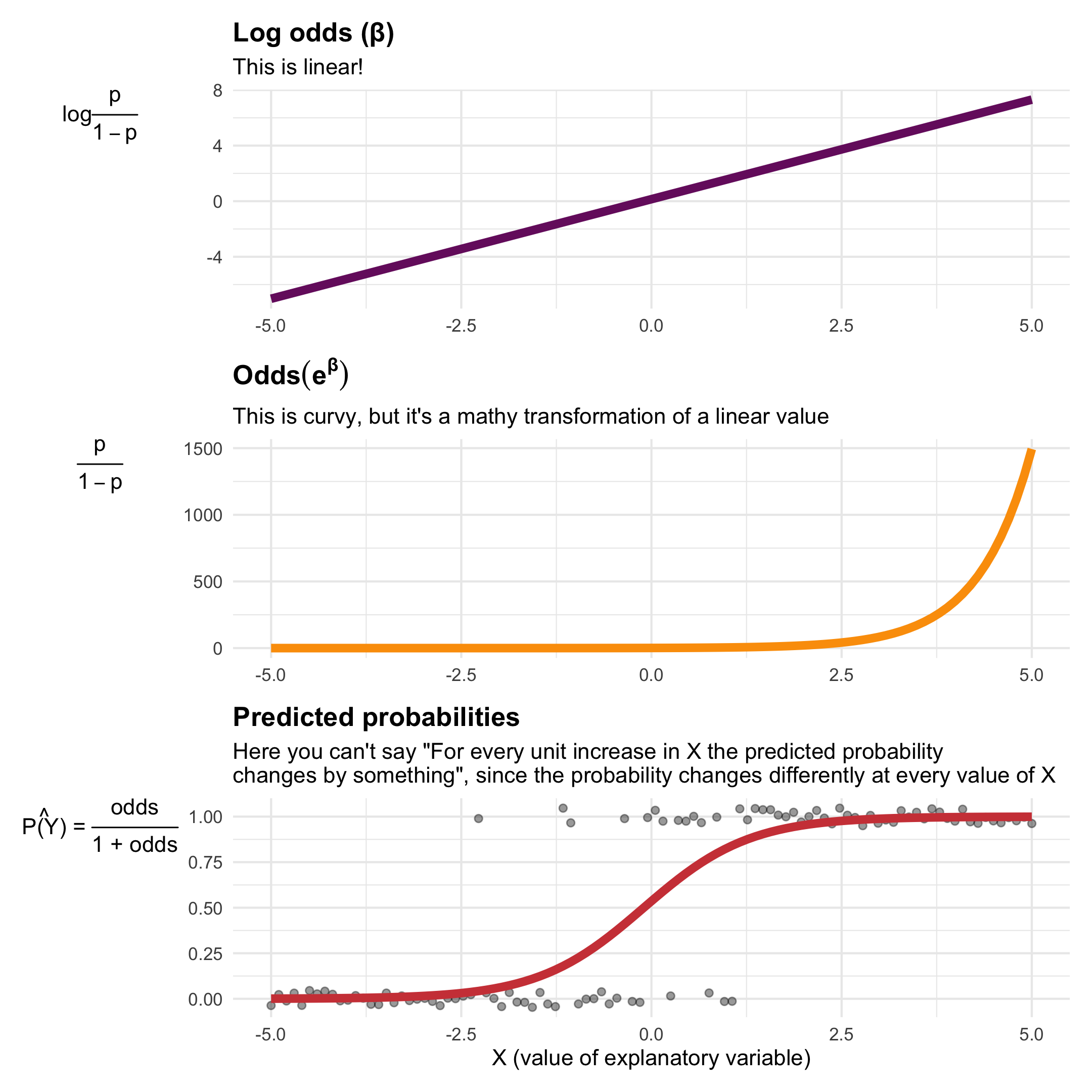
Just one last little explanation for why we have to use weird log odds things. When working with binary outcomes, we’re dealing with probabilities, and we can create something called “odds” with probabilities. If there’s a 70% chance of something happening, there’s a 30% chance of it not happening. The ratio of those two probabilities is called “odds”: \(\frac{p}{1 - p} = \frac{0.7}{1 - 0.7} = \frac{0.7}{0.3} = 2.333\). Odds typically follow a curvy relationship—as you move up to higher levels of your explanatory variable, the odds get bigger faster. If you log these odds, though (\(\log \frac{p}{1 - p}\)), the relationship becomes linear, which means we can use regular old linear regression on probabilities. Magic!
You can see the relationship between log odds and odds ratios in the first two panels here (this is fake data where X ranges between -5 and 5, and Y is either 0 or 1; you can see the data points in the final panel as dots):
The coefficients from logistic regression are log odds because they come from model that creates that nice straight line in the first panel. Log odds are impossible to interpret, so we can unlog them (\(e^\beta\)) to turn them into odds ratios.
The bottom panel shows predicted probabilities. You can do one more mathematical transformation with the odds (\(\frac{p}{1 - p}\)) to generate a probability instead of odds: \(\frac{\text{odds}}{1 + \text{odds}}\). That is what a propensity score is.
BUT AGAIN I cannot stress enough how much you don’t need to worry about the inner mechanics of logistic regression for this class! If that went over your head, don’t worry! All we’re doing for IPW is using logistic regression to create propensity scores, and the code below shows how to do that. Behind the scenes you’re moving from log odds (they’re linear!) to odds (they’re interpretable-ish) to probabilities (they’re super interpretable!), but you don’t need to worry about that.
PHEW. With that little tangent into logistic regression done, we can now build a model to generate propensity scores (or predicted probabilities).
When we include variables in the model that generates propensity scores, we’re making adjustments and closing backdoors in the DAG, just like we did with matching. But unlike matching, we’re not throwing any data away! We’re just making some observations more important and others less important.
First we build a model that predicts net usage based on income, nighttime temperatures, and health (since those nodes are our confounders from the DAG):
Code
model_net <-glm(net ~ income + temperature + health,data = nets,family =binomial(link ="logit"))# We could look at these results if we wanted, but we don't need to for this class# tidy(model_net, exponentiate = TRUE)
We can then plug in the income, temperatures, and health for every row in our dataset and generate a predicted probability using this model:
Code
# augment_columns() handles the plugging in of values. You need to feed it the# name of the model and the name of the dataset you want to add the predictions# to. The type.predict = "response" argument makes it so the predictions are in# the 0-1 scale. If you don't include that, you'll get predictions in an# uninterpretable log odds scale.net_probabilities <-augment_columns(model_net, nets,type.predict ="response") %>%# The predictions are in a column named ".fitted", so we rename it hererename(propensity = .fitted)# Look at the first few rows of a few columnsnet_probabilities %>%select(id, net, income, temperature, health, propensity) %>%head()## # A tibble: 6 × 6## id net income temperature health propensity## <dbl> <lgl> <dbl> <dbl> <dbl> <dbl>## 1 1 TRUE 781 21.1 56 0.367## 2 2 FALSE 974 26.5 57 0.389## 3 3 FALSE 502 25.6 15 0.158## 4 4 TRUE 671 21.3 20 0.263## 5 5 FALSE 728 19.2 17 0.308## 6 6 FALSE 1050 25.3 48 0.429
The propensity scores are in the propensity column. Some people, like person 3, are unlikely to use nets (only a 15.8% chance) given their levels of income, temperature, and health. Others like person 6 have a higher probability (42.9%) since their income and health are higher. Neat.
Next we need to convert those propensity scores into inverse probability weights, which makes weird observations more important (i.e. people who had a high probability of using a net but didn’t, and vice versa). To do this, we follow this equation:
This equation will create weights that provide the average treatment effect (ATE), but there are other versions that let you find the average treatment effect on the treated (ATT), average treatment effect on the controls (ATC), and a bunch of others. You can find those equations here.
We’ll use mutate() to create a column for the inverse probability weight:
Code
net_ipw <- net_probabilities %>%mutate(ipw = (net_num / propensity) + ((1- net_num) / (1- propensity)))# Look at the first few rows of a few columnsnet_ipw %>%select(id, net, income, temperature, health, propensity, ipw) %>%head()## # A tibble: 6 × 7## id net income temperature health propensity ipw## <dbl> <lgl> <dbl> <dbl> <dbl> <dbl> <dbl>## 1 1 TRUE 781 21.1 56 0.367 2.72## 2 2 FALSE 974 26.5 57 0.389 1.64## 3 3 FALSE 502 25.6 15 0.158 1.19## 4 4 TRUE 671 21.3 20 0.263 3.81## 5 5 FALSE 728 19.2 17 0.308 1.44## 6 6 FALSE 1050 25.3 48 0.429 1.75
These first few rows have fairly low weights—those with low probabilities of using nets didn’t, while those with high probabilities did. But look at person 4! They only had a 26% chance of using a net and they did! That’s weird! They therefore have a higher inverse probability weight (3.81).
Step 2: Estimation
Now that we’ve generated inverse probability weights based on our confounders, we can run a model to find the causal effect of mosquito net usage on malaria risk. Again, we don’t need to include income, temperatures, or health in the model since we already used them when we created the propensity scores and weights:
Cool! After using the inverse probability weights, we find a -10.13 point causal effect. That’s a tiiiny bit off from the true value of 10, but not bad at all!
It’s important to check the values of your inverse probability weights. Sometimes they can get too big, like if someone had an income of 0 and the lowest possible health and lived in a snow field and yet still used a net. Having really really high IPW values can throw off estimation. To fix this, we can truncate weights at some lower level. There’s no universal rule of thumb for a good maximum weight—I’ve often seen 10 used. In our mosquito net data, no IPWs are higher than 10 (the max is exactly 10 with person 877), so we don’t need to worry about truncation.
If you did want to truncate, you’d do something like this (here we’re truncating at 8 instead of 10 so truncation actually does something):
Code
net_ipw <- net_ipw %>%# If the IPW is larger than 8, make it 8, otherwise use the current IPWmutate(ipw_truncated =ifelse(ipw >8, 8, ipw))model_ipw_truncated <-lm(malaria_risk ~ net,data = net_ipw,weights = ipw_truncated)tidy(model_ipw_truncated)## # A tibble: 2 × 5## term estimate std.error statistic p.value## <chr> <dbl> <dbl> <dbl> <dbl>## 1 (Intercept) 39.7 0.467 85.1 0 ## 2 netTRUE -10.2 0.656 -15.5 4.32e-51
Now the causal effect is -10.19, which is slightly lower and probably less accurate since we don’t really have any exceptional cases messing up our original IPW estimate.
Results from all the models
All done! We just used observational data to estimate causal effects. Causation without RCTs! Do you know how neat that is?!
Let’s compare all the ATEs that we just calculated:
Which one is right? In this case, because this is fake simulated data where I built in a 10 point effect, we can see which of these models gets the closest: here, the non-truncated IPW model wins. But that won’t always be the case. Both matching and IPW work well for closing backdoors and adjusting for confounders. In real life, you won’t know the true value, so try multiple ways.
jklol we’re not all done! Fixing IPW standard errors and confidence intervals
So actually I lied. We’re not all done yet. The IPW model that we ran gives us the correct point estimate (-10.13), but the standard error and confidence intervals are wrong. There’s uncertainty inherent in the estimation of the propensity scores and inverse probability weights, and that uncertainty doesn’t get passed on to the outcome model, thus resulting in underestimation of the standard error and overly narrow confidence intervals.
We can actually see underestimation in the side-by-side table of results up above: the IPW model’s standard error is 0.66, resulting in a confidence interval of [-11.42, -8.84], while the matching + weights model has a similar point estimate (-10.48) but a bigger standard error (0.76) and a wider confidence interval of [-11.98, -8.98].
The (arguably) most accurate way to include the uncertainty of the treatment model in the outcome model is to rely on brute force simulation (just like we did with Zilch!) to estimate the causal effect for lots and lots of different models and then combine them into a single value. With our Zilch simulations, we rerolled our dice thousands of times. With this data, however, we can’t go out and collect new samples of mosquito net use—we only have one set of data to work with.
So instead of collecting new data, we cheat.
We’ll use a super-wild-that-this-even-works statistical procedure called bootstrapping to create random modified versions of our dataset.
Intuition behind bootstrapping
To help with the concept of bootstrapping, imagine we have 10 rows of data that looks like this:
ID
Name
Cookies
1
Mariah
3
2
Hibaq
4
3
Manaahil
3
4
Kadi
2
5
Ghaazi
4
6
Thanaa
1
7
Emilio
2
8
Jawhar
4
9
Iyaad
3
10
Sabiyya
1
With bootstrapping, we randomly select 10 rows from this original dataset to create a new dataset, and we do this some large number of times (like 1,000). Importantly, we randomly select rows with replacement, so that each random bootstrapped dataset will have some repeated rows. See this, for instance, where Sabiyya appears three times in the first bootstrapped dataset, Mariah twice in the second, and Kadi three times in the thousandth.
Original data
Bootstrap sample #1
Bootstrap sample #2
Bootstrap sample #1000
ID
Name
Cookies
ID
Name
Cookies
ID
Name
Cookies
…
ID
Name
Cookies
1
Mariah
3
2
Hibaq
4
1
Mariah
3
2
Hibaq
4
2
Hibaq
4
3
Manaahil
3
1
Mariah
3
4
Kadi
2
3
Manaahil
3
6
Thanaa
1
3
Manaahil
3
4
Kadi
2
4
Kadi
2
6
Thanaa
1
4
Kadi
2
4
Kadi
2
5
Ghaazi
4
7
Emilio
2
6
Thanaa
1
6
Thanaa
1
6
Thanaa
1
8
Jawhar
4
7
Emilio
2
7
Emilio
2
7
Emilio
2
8
Jawhar
4
8
Jawhar
4
8
Jawhar
4
8
Jawhar
4
10
Sabiyya
1
8
Jawhar
4
9
Iyaad
3
9
Iyaad
3
10
Sabiyya
1
9
Iyaad
3
9
Iyaad
3
10
Sabiyya
1
10
Sabiyya
1
10
Sabiyya
1
10
Sabiyya
1
We can then work with each of the bootstrapped datasets. If we wanted to find the standard error and confidence interval for the number of cookies eaten, we would calculate the average and standard error within each dataset, then combine all these averages into a single number or range.
The rsample package in R makes this easy to do by automatically splitting the dataset into random subsets for us. We can then create a function that R will run on each of the random datasets. Finally, we can collapse all the results into a single value. Here’s a quick example using the simulated cookie data from above.
We’ll use bootstrapping to find three specific numbers, or estimands:
The average number of cookies eaten
The standard error of the number of cookies eaten
The confidence interval of the number of cookies eaten
First we’ll use tribble() to make up a little toy dataset:
Next we’ll create a function that will calculate the stuff we’re interested in. This function will run an intercept-only model for each of the datasets we feed it. It feels weird to use a regression model to just get an average, but at its core regression is really just fancy averages. By using lm() and tidy(), we can automatically find averages, standard errors, and confidence intervals without extra math. Just go with it.
Now we can do some actual boostrapping. We’ll use the bootstraps() function from the rsample library to divide our dataset into 500 random datasets
Code
library(rsample)# apparent = TRUE includes the actual dataset as the final 501st row. It's# necessary for the fancy automatic confidence interval calculations that we'll# do laterset.seed(1234)cookie_samples <-bootstraps(cookie_data, times =500, apparent =TRUE)cookie_samples## # Bootstrap sampling with apparent sample ## # A tibble: 501 × 2## splits id ## <list> <chr> ## 1 <split [10/3]> Bootstrap001## 2 <split [10/5]> Bootstrap002## 3 <split [10/2]> Bootstrap003## 4 <split [10/2]> Bootstrap004## 5 <split [10/4]> Bootstrap005## 6 <split [10/5]> Bootstrap006## 7 <split [10/2]> Bootstrap007## 8 <split [10/2]> Bootstrap008## 9 <split [10/3]> Bootstrap009## 10 <split [10/3]> Bootstrap010## # … with 491 more rows
That new splits column contains a bunch of information about which rows are included in the new random dataset. If you’re curious about which rows are in which data, you can drill down into the boot_split object, but you don’t need to. For instance, here are the IDs included in the 5th simulated dataset. Neat.
Now that everything is split up and we have 500 datasets, we’ll apply our find_stats() function to each of the datasets using the map() function from the purrr package:
This looks a little weird. We have a new column named stats, but it doesn’t contain single numbers. Instead, each cell contains a whole dataset! Each cell holds the tidied results from the intercept-only regression model. We can look at the first nested dataset of results like this:
That looks like a regular table of regression results, with columns for the coefficient name, the estimate, standard error, p-value, and other things.
But all those numbers are nested inside datasets inside cells, which makes it really hard to work with. We can use the unnest() function to extract everything into regular columns:
2. The standard error of the number of cookies eaten
Finding the standard error is a little trickier. It’s tempting to just find the average of the std.error column like we did for the estimate column, but for mathy reasons this is wrong. Instead we need to use special equations developed by Donald Rubin (1987), often nicknamed “Rubin’s Rules”, that aggregate multiple standard errors into a single value in a way that adjusts for differences in those values. You can see simplified definitions of all of Rubin’s Rules here.
We didn’t need to do anything fancy for the average number of cookies eaten because Rubin’s Rules say that you can just take the average of an estimate to collapse (or pool) the results to a single number. Pooling standard errors requires a special formula that involves both the standard errors and the estimated averages:
3. The confidence interval of the number of cookies eaten
Since confidence intervals are based on standard errors, we also can’t just take the average of the conf.low and conf.high columns in each of the bootstrapped results. We have to follow one of the special Rubin’s Rules to collapse the confidence intervals into single values. We could do this by hand by multiplying the standard error we found above by 1.96 and −1.96 and then add those to the average, but that’s a lot of work. Instead, we can use the int_t() function from rsample to do this automatically.
The .lower and .upper columns here show the confidence interval, while the .estimate column shows the average number of cookies eaten.
Code
# (Note that we have to use this function on the dataset of nested results, not# the one where we unnested everything.)cookie_bootstrap_stats %>%int_t(stats)## # A tibble: 1 × 6## term .lower .estimate .upper .alpha .method ## <chr> <dbl> <dbl> <dbl> <dbl> <chr> ## 1 (Intercept) 1.54 2.71 3.47 0.05 student-t
Distributions and percentiles
int_t() technically works by using the critical values of the t-distribution at 2.5% and 97.5% (thus making a 95% confidence interval). If you want to be fancy and not assume that the distribution of the estimate follows a t distribution, you can use int_pctl() instead, which just finds the observed values at the 2.5% and 97.% percentiles without making any distributional assumptions.
Bootstrapping with inverse probability weighting
Phew. Now that we get the rough intuition behind bootstrapping (create a bunch of random copies of the original data, calculate stuff on each copy, and collapse those results into a single number), we can go through the same process with inverse probability weighting to get standard errors and confidence intervals that incorporate the uncertainty of the propensity score model and are thus more accurate.
We need to create a little function that will run the propensity score model, generate inverse probability weights, and run the outcome model for each of our random datasets. These are all the steps we ran earlier in the example, just in one function, running on a dataset named current_data:
Code
fit_one_ipw <-function(split) {# Work with just a sampled subset of the full data current_data <-analysis(split)# Fit propensity score model model_net <-glm(net ~ income + temperature + health,data = current_data,family =binomial(link ="logit"))# Calculate inverse probability weights net_ipw <-augment_columns(model_net, current_data,type.predict ="response") %>%mutate(ipw = (net_num / .fitted) + ((1- net_num) / (1- .fitted)))# Fit outcome model with IPWs model_outcome <-lm(malaria_risk ~ net,data = net_ipw,weights = ipw)# Return a tidied version of the model resultsreturn(tidy(model_outcome))}
With that function, we can take our nets data and make 1000 random copies of it. We’ll then apply our neat fit_one_ipw() function to each of the datasets
If we unnest that column, we can see the actual results. Notice how there are 2,002 rows here instead of 1,001—each dataset has two values in it: the intercept and the coefficient for netTRUE.
The average net effect (or point estimate) of -10.12 is the same as what we found with the regular lm() model from before, but the standard error is now bigger (0.78) and more like the standard error we got with matching + weights (0.76).
Finally we can find the confidence interval for the estimate with int_t():
That’s the correct result. Using inverse probability weighting, using a mosquito net causes a -10.12 point reduction in malaria risk scores, with a standard error of 0.78 and a 95% confidence interval of [-10.96, -9.34].
Source Code
---title: "Matching and inverse probability weighting"---```{r setup, include=FALSE}knitr::opts_chunk$set(fig.width =6, fig.asp =0.618, fig.align ="center",fig.retina =3, out.width ="75%", collapse =TRUE)set.seed(1234)options("digits"=2, "width"=150)options(dplyr.summarise.inform =FALSE)```## Video walk-throughIf you want to follow along with this example, you can download the data below:- [{{< fa table >}} `mosquito_nets.csv`](/files/data/generated_data/mosquito_nets.csv)```{r show-youtube-list, echo=FALSE, results="asis"}source(here::here("R", "youtube-playlist.R"))playlist_id <-"PLS6tnpTr39sGv7RPHEZy8CDSdCliQOW8K"video_details <- tibble::tribble(~youtube_id, ~title,"OoYgOwxUQgI", "Drawing a DAG","_FNrfFe99R0", "Creating an RStudio project","7NW7GbO44BY", "Naive (and wrong!) estimate","uGwSRnET8Sg", "Matching","CKm1rZlAwuA", "Inverse probability weighting")youtube_list(video_details, playlist_id, example =TRUE)```## Program backgroundLet's revisit the mosquito net program we used in [the DAG example](/example/dags.qmd). Here, researchers are interested in whether using mosquito nets decreases an individual's risk of contracting malaria. They have collected data from 1,752 households in an unnamed country and have variables related to environmental factors, individual health, and household characteristics. The data is **not experimental**—researchers have no control over who uses mosquito nets, and individual households make their own choices over whether to apply for free nets or buy their own nets, as well as whether they use the nets if they have them.The CSV file contains the following columns:- Malaria risk (`malaria_risk`): The likelihood that someone in the household will be infected with malaria. Measured on a scale of 0–100, with higher values indicating higher risk.- Mosquito net (`net` and `net_num`): A binary variable indicating if the household used mosquito nets.- Eligible for program (`eligible`): A binary variable indicating if the household is eligible for the free net program.- Income (`income`): The household's monthly income, in US dollars.- Nighttime temperatures (`temperature`): The average temperature at night, in Celsius.- Health (`health`): Self-reported healthiness in the household. Measured on a scale of 0–100, with higher values indicating better health.- Number in household (`household`): Number of people living in the household.- Insecticide resistance (`resistance`): Some strains of mosquitoes are more resistant to insecticide and thus pose a higher risk of infecting people with malaria. This is measured on a scale of 0–100, with higher values indicating higher resistance.## Our goal**Our goal in this example is to estimate the *causal* effect of bed net usage on malaria risk using *only* observational data.** This was not an RCT, so it might seem a little sketchy to make claims of causality. But if we can draw a correct DAG and adjust for the correct nodes, we can isolate the net → malaria relationship and talk about causality.Because this is simulated data, we know the true causal effect of the net program because I built it into the data. **The true average treatment effect (ATE) is -10.** Using a mosquito net *causes* the risk of malaria to decrease by 10 points, on average.Let's see if we can find that 10 point effect!## Load data and librariesFirst, let's download the dataset (if you haven't already), put in a folder named `data`, and load it:- [{{< fa table >}} `mosquito_nets.csv`](/files/data/generated_data/mosquito_nets.csv)```{r load-libraries, message=FALSE, warning=FALSE}library(tidyverse) # ggplot(), %>%, mutate(), and friendslibrary(ggdag) # Make DAGslibrary(dagitty) # Do DAG logic with Rlibrary(broom) # Convert models to data frameslibrary(MatchIt) # Match thingslibrary(modelsummary) # Make side-by-side regression tablesset.seed(1234) # Make all random draws reproducible``````{r load-data-fake, eval=FALSE}# Load the data.# It'd be a good idea to click on the "nets" object in the Environment panel in# RStudio to see what the data looks like after you load itnets <-read_csv("data/mosquito_nets.csv")``````{r load-data-real, include=FALSE}nets <-read_csv(here::here("files", "data", "generated_data", "mosquito_nets.csv"))```## DAG and adjustment setsBefore running any models, we need to find what we need to adjust for. Recall from [the DAG example](/example/dags.qmd) that we built this causal model to show the data generating process behind the relationship between mosquito net usage and malaria risk:```{r build-mosquito-dag, fig.width=8}mosquito_dag <-dagify( malaria_risk ~ net + income + health + temperature + resistance, net ~ income + health + temperature + eligible + household, eligible ~ income + household, health ~ income,exposure ="net",outcome ="malaria_risk",coords =list(x =c(malaria_risk =7, net =3, income =4, health =5,temperature =6, resistance =8.5, eligible =2, household =1),y =c(malaria_risk =2, net =2, income =3, health =1,temperature =3, resistance =2, eligible =3, household =2)),labels =c(malaria_risk ="Risk of malaria", net ="Mosquito net", income ="Income",health ="Health", temperature ="Nighttime temperatures",resistance ="Insecticide resistance",eligible ="Eligible for program", household ="Number in household"))ggdag_status(mosquito_dag, use_labels ="label", text =FALSE) +guides(fill ="none", color ="none") +# Disable the legendtheme_dag()```Following the logic of *do*-calculus, we can find all the nodes that confound the relationship between net usage and malaria risk, since those nodes open up backdoor paths and distort the causal effect we care about. We can either do this graphically by looking for any node that points to both net and malaria risk, or we can use R:```{r show-adjustment-sets}adjustmentSets(mosquito_dag)```Based on the relationships between all the nodes in the DAG, adjusting for health, income, and temperature is enough to close all backdoors and identify the relationship between net use and malaria risk.## Naive correlation-isn't-causation estimateFor fun, we can calculate the difference in average malaria risk for those who did/didn't use mosquito nets. This is most definitely *not* the actual causal effect—this is the "correlation is not causation" effect that doesn't account for any of the backdoors in the DAG.We can do this with a table (but then we have to do manual math to subtract the `FALSE` average from the `TRUE` average):```{r super-wrong-answer-table}nets %>%group_by(net) %>%summarize(number =n(),avg =mean(malaria_risk))```Or we can do it with regression:```{r super-wrong-answer}model_wrong <-lm(malaria_risk ~ net, data = nets)tidy(model_wrong)```According to this estimate, using a mosquito net is associated with a `r tidy(model_wrong) %>% filter(term == "netTRUE") %>% pull(estimate) %>% abs()` point decrease in malaria risk, on average. We can't legally talk about this as a causal effect though—there are confounding variables to deal with.## MatchingWe can use matching techniques to pair up similar observations and make the unconfoundedness assumption—that if we see two observations that are pretty much identical, and one used a net and one didn't, the choice to use a net was random.Because we know from the DAG that income, nighttime temperatures, and health help cause both net use and malaria risk (and confound that relationship!), we'll try to find observations with similar values of income, temperatures, and health that both used and didn't use nets.We can use the `matchit()` function from the **MatchIt** R package to match points based on Mahalanobis distance. There are lots of other options available—see [the online documentation](http://gking.harvard.edu/matchit) for details.We can include the `replace = TRUE` option to make it so that points that have been matched already can be matched again (that is, we're not forcing a one-to-one matching; we have one-to-many matching instead).### Step 1: Preprocess```{r matching}matched_data <-matchit(net ~ income + temperature + health,data = nets,method ="nearest",distance ="mahalanobis",replace =TRUE)summary(matched_data)```Here we can see that all 681 of the net users were paired with similar-looking non-users (439 of them). 632 people weren't matched and will get discarded. If you're curious, you can see which treated rows got matched to which control rows by running `matched_data$match.matrix`.We can create a new data frame of those matches with `match.data()`:```{r matched-data}matched_data_for_real <-match.data(matched_data)```### Step 2: EstimationNow that the data has been matched, it should work better for modeling. Also, because we used income, temperatures, and health in the matching process, we've adjusted for those DAG nodes and have closed those backdoors, so our model can be pretty simple here:```{r matching-model}model_matched <-lm(malaria_risk ~ net,data = matched_data_for_real)tidy(model_matched)```The `r tidy(model_matched) %>% filter(term == "netTRUE") %>% pull(estimate) %>% abs()` point decrease here is better than the naive estimate, but it's not the true 10 point causal effect (that I built in to the data). Perhaps that's because the matches aren't great, or maybe we threw away too much data. There are a host of diagnostics you can look at to see how well things are matched (check [the documentation for **MatchIt**](http://gking.harvard.edu/matchit) for examples.)Actually, the most likely culprit for the incorrect estimate is that there's some imbalance in the data. Because we set `replace = TRUE`, we did _not_ do 1:1 matching—untreated observations were paired with more than one treated observation. As a result, the multiply-matched observations are getting overcounted and have too much importance in the model. Fortunately, `matchit()` provides us with a column called `weights` that allows us to scale down the overmatched observations when running the model. **Importantly,** these weights have nothing to do with causal inference or backdoors or inverse probability weighting—their only purpose is to help scale down the imbalance arising from overmatching. If you use `replace = FALSE` and enforce 1:1 matching, the whole `weights` column will just be 1.We can incorporate those weights into the model and get a more accurate estimate:```{r matching-model-weight}model_matched_wts <-lm(malaria_risk ~ net,data = matched_data_for_real,weights = weights)tidy(model_matched_wts)```After weighting to account for under- and over-matching, we find a `r tidy(model_matched_wts) %>% filter(term == "netTRUE") %>% pull(estimate)` point causal effect. That's much better than any of the other estimates we've tried so far! The reason it's accurate is because we've closed the confounding backdoors and isolated the arrow between net use and malaria risk.## Inverse probability weightingOne potential downside to matching is that you generally have to throw away a sizable chunk of your data—anything that's unmatched doesn't get included in the final matched data.An alternative approach to matching is to assign every observation some probability of receiving treatment, and then weight each observation by its inverse probability—observations that are predicted to get treatment and then don't, or observations that are predicted to not get treatment and then do will receive more weight than the observations that get/don't get treatment as predicted.Generating these inverse probability weights requires a two step process: (1) we first generate propensity scores, or the probability of receiving treatment, and then (2) we use a special formula to convert those propensity scores into weights. Once we have inverse probability weights weights, we can incorporate them into our regression model.### Oversimplified crash course in logistic regressionThere are many ways to generate propensity scores (like logistic regression, probit regression, and even machine learning techniques like random forests and neural networks), but logistic regression is probably the most common method.The complete technical details of logistic regression are beyond the scope of this class, but if you're curious you should check out [this highly accessible tutorial](https://uc-r.github.io/logistic_regression).All you really need to know is that the outcome variable in logistic regression models must be binary, and the explanatory variables you include in the model help explain the variation in the likelihood of your binary outcome. The Y (or outcome) in logistic regression is a logged ratio of probabilities, which forces the model's output to be in a 0-1 range:$$\log \frac{p_y}{p_{1-y}} = \beta_0 + \beta_1 x_1 + \beta_2 x_2 + \dots + \beta_n x_n + \epsilon$$Here's what it looks like visually. Because net usage is a binary outcome, there are lines of observations at 0 and 1 along the y axis. The blue curvy line here shows the output of a logistic regression model—people with low income have a low likelihood of using a net, while those with high income are far more likely to do so.I also included a red line showing the results from a regular old `lm()` OLS model. It follows the blue line fairly well for a while, but predicts negative probabilities if you use lower values of income, like less than 250. For strange historical and mathy reasons, many economists like using OLS on binary outcomes (they even have a fancy name for it: linear probability models (LPMs)), but I'm partial to logistic regression since it doesn't generate probabilities greater than 100% or less than 0%. (BUT DON'T EVER COMPLAIN ABOUT LPMs ONLINE. You'll start battles between economists and other social scientists. 🤣)```{r net-income-logit, warning=FALSE, message=FALSE}ggplot(nets, aes(x = income, y = net_num)) +geom_point(alpha =0.1) +geom_smooth(method ="lm", color ="red", size =0.5) +geom_smooth(method ="glm", method.args =list(family =binomial(link ="logit"))) +labs(x ="Income", y ="Probability of using a net")```The coefficients from a logistic regression model are interpreted differently than you're used to (and their interpretations can be controversial!). Here's an example for the model in the graph above:```{r logit-output}# Notice how we use glm() instead of lm(). The "g" stands for "generalized"# linear model. We have to specify a family in any glm() model. You can# technically run a regular OLS model (like you do with lm()) if you use# glm(y ~ x1 + x2, family = gaussian(link = "identity")), but people rarely do that.## To use logistic regression, you have to specify a binomial/logit family like so:# family = binomial(link = "logit")model_logit <-glm(net ~ income + temperature + health,data = nets,family =binomial(link ="logit"))tidy(model_logit)```The coefficients here aren't normal numbers—they're called "log odds" and represent the change in the logged odds as you move explanatory variables up. For instance, here the logged odds of using a net increase by 0.00688 for every one point increase in your health score. But what do logged odds even mean?! Nobody knows.You can make these coefficients slightly more interpretable by unlogging them and creating something called an "odds ratio." These coefficients were logged with a natural log, so you unlog them by raising $e$ to the power of the coefficient. The odds ratio for temperature is $e^{-0.0589}$, or `r exp(-0.0589)`. Odds ratios get interpreted a little differently than regular model coefficients. Odds ratios are all centered around 1—values above 1 mean that there's an increase in the likelihood of the outcome, while values below 1 mean that there's a decrease in the likelihood of the outcome.Our nighttime temperature odds ratio here is 0.94, which is 0.06 below 1, which means we can say that for every one point increase in nighttime temperatures, a person is 6% less likely to use a net. If the coefficient was something like 1.34, we could say that they'd be *34% more likely* to use a net; if it was something like 5.02 we could say that they'd be *5 times more* likely to use a net; if it was something like 0.1, we could say that they're *90% less likely* to use a net.You can make R exponentiate the coefficients automatically by including `exponentiate = TRUE` in `tidy()`:```{r logit-output-exp}tidy(model_logit, exponentiate =TRUE)```**BUT AGAIN** this goes beyond the scope of this class! Just know that when you build a logistic regression model, you're using explanatory variables to predict the probability of an outcome.Just one last little explanation for why we have to use weird log odds things. When working with binary outcomes, we're dealing with probabilities, and we can create something called "odds" with probabilities. If there's a 70% chance of something happening, there's a 30% chance of it not happening. The ratio of those two probabilities is called "odds": $\frac{p}{1 - p} = \frac{0.7}{1 - 0.7} = \frac{0.7}{0.3} = 2.333$. Odds typically follow a curvy relationship—as you move up to higher levels of your explanatory variable, the odds get bigger faster. If you log these odds, though ($\log \frac{p}{1 - p}$), the relationship becomes linear, which means we can use regular old linear regression on probabilities. Magic!You can see the relationship between log odds and odds ratios in the first two panels here (this is fake data where X ranges between -5 and 5, and Y is either 0 or 1; you can see the data points in the final panel as dots):```{r odds-probs, echo=FALSE, fig.width=7.5, fig.height=4.75, fig.asp=1, out.width="80%"}library(latex2exp)library(patchwork)# Using TeX() wipes out theming for whatever reason, so this manually bolds stuffbolded_tex <-function(str) parse(text =paste0("bold(", TeX(str), ")"))set.seed(1234)logit_df <-tibble(x =seq(-5, 5, length.out =100)) %>%mutate(p =1/ (1+exp(-x))) %>%mutate(y =rbinom(n(), size =1, prob = p))example_logit <-glm(y ~ x, data = logit_df, family =binomial(link ="logit"))logit_df_odds <-augment_columns(example_logit, logit_df,type.predict =c("link")) %>%rename(log_odds = .fitted) %>%mutate(odds_ratio =exp(log_odds))logit_df_probs <-augment_columns(example_logit, logit_df,type.predict =c("response")) %>%rename(pred_prob = .fitted)plot_log_odds <-ggplot(logit_df_odds, aes(x = x, y = log_odds)) +geom_path(color ="#771C6D", size =2) +labs(title ="Log odds (β)",subtitle ="This is linear!",x =NULL,y =TeX("$log \\frac{p}{1 - p}$")) +theme_minimal() +theme(plot.title =element_text(face ="bold"),axis.title.y =element_text(angle =0, hjust =0))plot_odds <-ggplot(logit_df_odds, aes(x = x, y = odds_ratio)) +geom_path(color ="#FB9E07", size =2) +labs(title =bolded_tex("Odds (e^β)"),subtitle ="This is curvy, but it's a mathy transformation of a linear value",x =NULL,y =TeX("$\\frac{p}{1 - p}$")) +theme_minimal() +theme(plot.title =element_text(face ="bold"),axis.title.y =element_text(angle =0, hjust =0))plot_probs <-ggplot(logit_df_probs, aes(x = x, y = pred_prob)) +geom_point(aes(y = y), alpha =0.4, position =position_jitter(width =0, height =0.05)) +geom_path(color ="#CF4446", size =2) +labs(title ="Predicted probabilities",subtitle ='Here you can\'t say "For every unit increase in X the predicted probability\nchanges by something", since the probability changes differently at every value of X',x ="X (value of explanatory variable)",y =TeX("\\hat{P(Y)} = \\frac{odds}{1 + odds}")) +theme_minimal() +theme(plot.title =element_text(face ="bold"),axis.title.y =element_text(angle =0, hjust =0))plot_log_odds / plot_odds / plot_probs```The coefficients from logistic regression are log odds because they come from model that creates that nice straight line in the first panel. Log odds are impossible to interpret, so we can unlog them ($e^\beta$) to turn them into odds ratios.The bottom panel shows predicted probabilities. You can do one more mathematical transformation with the odds ($\frac{p}{1 - p}$) to generate a probability instead of odds: $\frac{\text{odds}}{1 + \text{odds}}$. ***That*** is what a propensity score is.**BUT AGAIN I cannot stress enough how much you don't need to worry about the inner mechanics of logistic regression for this class!** If that went over your head, don't worry! All we're doing for IPW is using logistic regression to create propensity scores, and the code below shows how to do that. Behind the scenes you're moving from log odds (they're linear!) to odds (they're interpretable-ish) to probabilities (they're super interpretable!), but you don't need to worry about that.If you're confused, [watch this TikTok for consolation](https://twitter.com/ChelseaParlett/status/1304436259926896640).### Step 1: Generate propensity scoresPHEW. With that little tangent into logistic regression done, we can now build a model to generate propensity scores (or predicted probabilities).When we include variables in the model that generates propensity scores, we're making adjustments and closing backdoors in the DAG, just like we did with matching. But unlike matching, we're not throwing any data away! We're just making some observations more important and others less important.First we build a model that predicts net usage based on income, nighttime temperatures, and health (since those nodes are our confounders from the DAG):```{r make-net-logit-model}model_net <-glm(net ~ income + temperature + health,data = nets,family =binomial(link ="logit"))# We could look at these results if we wanted, but we don't need to for this class# tidy(model_net, exponentiate = TRUE)```We can then plug in the income, temperatures, and health for every row in our dataset and generate a predicted probability using this model:```{r make-p-scores}# augment_columns() handles the plugging in of values. You need to feed it the# name of the model and the name of the dataset you want to add the predictions# to. The type.predict = "response" argument makes it so the predictions are in# the 0-1 scale. If you don't include that, you'll get predictions in an# uninterpretable log odds scale.net_probabilities <-augment_columns(model_net, nets,type.predict ="response") %>%# The predictions are in a column named ".fitted", so we rename it hererename(propensity = .fitted)# Look at the first few rows of a few columnsnet_probabilities %>%select(id, net, income, temperature, health, propensity) %>%head()```The propensity scores are in the `propensity` column. Some people, like person 3, are unlikely to use nets (only a 15.8% chance) given their levels of income, temperature, and health. Others like person 6 have a higher probability (42.9%) since their income and health are higher. Neat.Next we need to convert those propensity scores into inverse probability weights, which makes weird observations more important (i.e. people who had a high probability of using a net but didn't, and vice versa). To do this, we follow this equation:$$\frac{\text{Treatment}}{\text{Propensity}} + \frac{1 - \text{Treatment}}{1 - \text{Propensity}}$$This equation will create weights that provide the average treatment effect (ATE), but there are other versions that let you find the average treatment effect on the treated (ATT), average treatment effect on the controls (ATC), and a bunch of others. [You can find those equations here](https://livefreeordichotomize.com/2019/01/17/understanding-propensity-score-weighting/#how-do-we-incorporate-a-propensity-score-in-a-weight).We'll use `mutate()` to create a column for the inverse probability weight:```{r make-ipw}net_ipw <- net_probabilities %>%mutate(ipw = (net_num / propensity) + ((1- net_num) / (1- propensity)))# Look at the first few rows of a few columnsnet_ipw %>%select(id, net, income, temperature, health, propensity, ipw) %>%head()```These first few rows have fairly low weights—those with low probabilities of using nets didn't, while those with high probabilities did. But look at person 4! They only had a 26% chance of using a net and they *did*! That's weird! They therefore have a higher inverse probability weight (3.81).### Step 2: EstimationNow that we've generated inverse probability weights based on our confounders, we can run a model to find the causal effect of mosquito net usage on malaria risk. Again, we don't need to include income, temperatures, or health in the model since we already used them when we created the propensity scores and weights:```{r ipw-model}model_ipw <-lm(malaria_risk ~ net,data = net_ipw,weights = ipw)tidy(model_ipw)```Cool! After using the inverse probability weights, we find a `r tidy(model_ipw) %>% filter(term == "netTRUE") %>% pull(estimate)` point causal effect. That's a tiiiny bit off from the true value of 10, but not bad at all!It's important to check the values of your inverse probability weights. Sometimes they can get too big, like if someone had an income of 0 and the lowest possible health and lived in a snow field *and yet* still used a net. Having really really high IPW values can throw off estimation. To fix this, we can truncate weights at some lower level. There's no universal rule of thumb for a good maximum weight—I've often seen 10 used. In our mosquito net data, no IPWs are higher than 10 (the max is exactly 10 with person 877), so we don't need to worry about truncation.If you did want to truncate, you'd do something like this (here we're truncating at 8 instead of 10 so truncation actually does something):```{r ipw-truncated}net_ipw <- net_ipw %>%# If the IPW is larger than 8, make it 8, otherwise use the current IPWmutate(ipw_truncated =ifelse(ipw >8, 8, ipw))model_ipw_truncated <-lm(malaria_risk ~ net,data = net_ipw,weights = ipw_truncated)tidy(model_ipw_truncated)```Now the causal effect is `r tidy(model_ipw_truncated) %>% filter(term == "netTRUE") %>% pull(estimate)`, which is slightly lower and probably less accurate since we don't really have any exceptional cases messing up our original IPW estimate.## Results from all the modelsAll done! We just used observational data to estimate causal effects. Causation without RCTs! Do you know how neat that is?!Let's compare all the ATEs that we just calculated:```{r all-models-together-fake, eval=FALSE}modelsummary(list("Naive"= model_wrong,"Matched"= model_matched, "Matched + weights"= model_matched_wts,"IPW"= model_ipw, "IPW truncated at 8"= model_ipw_truncated))``````{r all-models-together-real, echo=FALSE, warning=FALSE}gm <- modelsummary::gof_map %>%mutate(omit =!(raw %in%c("nobs", "r.squared", "adj.r.squared")))modelsummary(list("Naive"= model_wrong,"Matched"= model_matched, "Matched + weights"= model_matched_wts,"IPW"= model_ipw, "IPW truncated at 8"= model_ipw_truncated),gof_map = gm, stars =TRUE) %>% kableExtra::row_spec(3, background ="#F6D645") %>% kableExtra::row_spec(4, extra_css ="border-bottom: 1px solid")```Which one is right? In this case, because this is fake simulated data where I built in a 10 point effect, we can see which of these models gets the closest: here, the non-truncated IPW model wins. But that won't always be the case. Both matching and IPW work well for closing backdoors and adjusting for confounders. In real life, you won't know the true value, so try multiple ways.## jklol we're not all done! Fixing IPW standard errors and confidence intervals```{r extract-ipw-results, include=FALSE}ipw_results <- model_ipw %>%tidy(conf.int =TRUE) %>%mutate(term = janitor::make_clean_names(term)) %>%mutate(conf = glue::glue("[{round(conf.low, 2)}, {round(conf.high, 2)}]")) %>%split(~term)matching_results <- model_matched_wts %>%tidy(conf.int =TRUE) %>%mutate(term = janitor::make_clean_names(term)) %>%mutate(conf = glue::glue("[{round(conf.low, 2)}, {round(conf.high, 2)}]")) %>%split(~term)```So actually I lied. We're *not* all done yet. The IPW model that we ran gives us the correct point estimate (`r round(ipw_results$net_true$estimate, 2)`), but the standard error and confidence intervals are wrong. There's uncertainty inherent in the estimation of the propensity scores and inverse probability weights, and that uncertainty doesn't get passed on to the outcome model, thus resulting in underestimation of the standard error and overly narrow confidence intervals. We can actually see underestimation in the side-by-side table of results up above: the IPW model's standard error is `r round(ipw_results$net_true$std.error, 2)`, resulting in a confidence interval of `r ipw_results$net_true$conf`, while the matching + weights model has a similar point estimate (`r round(matching_results$net_true$estimate, 2)`) but a bigger standard error (`r round(matching_results$net_true$std.error, 2)`) and a wider confidence interval of `r matching_results$net_true$conf`.The (arguably) most accurate way to include the uncertainty of the treatment model in the outcome model is to rely on brute force simulation (just like we [did with Zilch!](/resource/zilch.qmd)) to estimate the causal effect for lots and lots of different models and then combine them into a single value. With our Zilch simulations, we rerolled our dice thousands of times. With this data, however, we can't go out and collect new samples of mosquito net use—we only have one set of data to work with.So instead of collecting new data, we cheat. We'll use a super-wild-that-this-even-works statistical procedure called [bootstrapping](https://en.wikipedia.org/wiki/Bootstrapping_(statistics)) to create random modified versions of our dataset.### Intuition behind bootstrappingTo help with the concept of bootstrapping, imagine we have 10 rows of data that looks like this:```{r generate-boot-example, echo=FALSE, warning=FALSE, message=FALSE}library(randomNames)library(kableExtra)withr::with_seed(1234, { example_data <-tibble(ID =1:10, Name =randomNames(n =10, which.names ="first")) %>%mutate(Cookies =rpois(n(), 3))})example_data %>%kbl() %>%kable_styling(full_width =FALSE)```With bootstrapping, we randomly select 10 rows from this original dataset to create a new dataset, and we do this some large number of times (like 1,000). Importantly, we randomly select rows *with replacement*, so that each random bootstrapped dataset will have some repeated rows. See this, for instance, where Sabiyya appears three times in the first bootstrapped dataset, Mariah twice in the second, and Kadi three times in the thousandth.```{r show-bootstrap-example, echo=FALSE, warning=FALSE, message=FALSE}set.seed(12345)boot1 <-tibble(ID =sample(example_data$ID, size =10, replace =TRUE)) %>%left_join(example_data, by ="ID") %>%arrange(ID)boot2 <-tibble(ID =sample(example_data$ID, size =10, replace =TRUE)) %>%left_join(example_data, by ="ID") %>%arrange(ID)boot3 <-tibble(ID =sample(example_data$ID, size =10, replace =TRUE)) %>%left_join(example_data, by ="ID") %>%arrange(ID)blank <-tibble(dots =rep("", 10))columns <-c(rep(c("ID", "Name", "Cookies"), times =3),"…",c("ID", "Name", "Cookies"))bind_cols(example_data, boot1, boot2, blank, boot3) %>%kbl(col.names = columns) %>%kable_styling() %>%add_header_above(c("Original data"=3, "Bootstrap sample #1"=3,"Bootstrap sample #2"=3, " "=1, "Bootstrap sample #1000"=3)) %>%column_spec(1:3, background ="#dedede")```We can then work with each of the bootstrapped datasets. If we wanted to find the standard error and confidence interval for the number of cookies eaten, we would calculate the average and standard error within each dataset, then combine all these averages into a single number or range.The `rsample` package in R makes this easy to do by automatically splitting the dataset into random subsets for us. We can then create a function that R will run on each of the random datasets. Finally, we can collapse all the results into a single value. Here's a quick example using the simulated cookie data from above. We'll use bootstrapping to find three specific numbers, or estimands:1. The average number of cookies eaten2. The standard error of the number of cookies eaten3. The confidence interval of the number of cookies eatenFirst we'll use `tribble()` to make up a little toy dataset:```{r make-cookie-data}cookie_data <-tribble(~ID, ~Name, ~Cookies,1, "Mariah", 3,2, "Hibaq", 4,3, "Manaahil", 3,4, "Kadi", 2,5, "Ghaazi", 4,6, "Thanaa", 1,7, "Emilio", 2,8, "Jawhar", 4,9, "Iyaad", 3,10, "Sabiyya", 1)cookie_data```Next we'll create a function that will calculate the stuff we're interested in. This function will run an intercept-only model for each of the datasets we feed it. It feels weird to use a regression model to just get an average, but at its core regression is really just fancy averages. By using `lm()` and `tidy()`, we can automatically find averages, standard errors, and confidence intervals without extra math. Just go with it. ```{r cookie-stats-function}find_stats <-function(splits) { current_data <-analysis(splits) cookie_model <-lm(Cookies ~1, data = current_data)return(tidy(cookie_model, conf.int =TRUE))}```Now we can do some actual boostrapping. We'll use the `bootstraps()` function from the `rsample` library to divide our dataset into 500 random datasets```{r split-cookies}library(rsample)# apparent = TRUE includes the actual dataset as the final 501st row. It's# necessary for the fancy automatic confidence interval calculations that we'll# do laterset.seed(1234)cookie_samples <-bootstraps(cookie_data, times =500, apparent =TRUE)cookie_samples```That new `splits` column contains a bunch of information about which rows are included in the new random dataset. If you're curious about which rows are in which data, you can drill down into the `boot_split` object, but you don't need to. For instance, here are the IDs included in the 5th simulated dataset. Neat.```{r show-some-ids}cookie_samples$splits[[5]]$in_id```Now that everything is split up and we have 500 datasets, we'll apply our `find_stats()` function to each of the datasets using the `map()` function from the `purrr` package:```{r find-cookie-stats, cache=TRUE}cookie_bootstrap_stats <- cookie_samples %>%mutate(stats =map(splits, find_stats))cookie_bootstrap_stats```This looks a little weird. We have a new column named `stats`, but it doesn't contain single numbers. Instead, each cell contains a whole dataset! Each cell holds the tidied results from the intercept-only regression model. We can look at the first nested dataset of results like this:```{r see-nested-cookie-df}cookie_bootstrap_stats$stats[[1]]```That looks like a regular table of regression results, with columns for the coefficient name, the estimate, standard error, p-value, and other things. But all those numbers are nested inside datasets inside cells, which makes it really hard to work with. We can use the `unnest()` function to extract everything into regular columns:```{r see-unnested-cookie-df, cache=TRUE}cookie_bootstrap_stats_unnested <- cookie_samples %>%mutate(stats =map(splits, find_stats)) %>%unnest(stats)cookie_bootstrap_stats_unnested```That's much more manageable! Now we can start finding the three estimands we're interested in.**1\. The average number of cookies eaten**To find the average number of cookies eaten, based on our boostrapped samples, we can calculate the average of the `estimate` column. That's it.```{r boot-avg-cookies}cookie_bootstrap_stats_unnested %>%summarize(avg_cookies =mean(estimate))```**2\. The standard error of the number of cookies eaten**Finding the standard error is a little trickier. It's tempting to just find the average of the `std.error` column like we did for the `estimate` column, but for mathy reasons this is wrong. Instead we need to use special equations developed by [Donald Rubin (1987)](https://onlinelibrary.wiley.com/doi/book/10.1002/9780470316696), often nicknamed "Rubin's Rules", that aggregate multiple standard errors into a single value in a way that adjusts for differences in those values. You can see [simplified definitions of all of Rubin's Rules here](https://bookdown.org/mwheymans/bookmi/rubins-rules.html).We didn't need to do anything fancy for the average number of cookies eaten because Rubin's Rules say that you can just take the average of an estimate to collapse (or pool) the results to a single number. Pooling standard errors requires a special formula that involves both the standard errors and the estimated averages:$$\sqrt{\operatorname{Average}(\text{standard errors}^2) + \operatorname{Variance}(\text{estimates})}$$Or in code:```{r rubin-se-example, eval=FALSE}sqrt(mean(errors^2) +var(estimates))```Let's do that with our cookie bootstraps:```{r boot-se-cookies}cookie_bootstrap_stats_unnested %>%summarize(avg_cookies =mean(estimate),avg_se =sqrt(mean(std.error^2) +var(estimate)))```**3\. The confidence interval of the number of cookies eaten**Since confidence intervals are based on standard errors, we also can't just take the average of the `conf.low` and `conf.high` columns in each of the bootstrapped results. We have to follow one of the special Rubin's Rules to collapse the confidence intervals into single values. We could do this by hand by multiplying the standard error we found above by 1.96 and −1.96 and then add those to the average, but that's a lot of work. Instead, we can use the `int_t()` function from `rsample` to do this automatically. The `.lower` and `.upper` columns here show the confidence interval, while the `.estimate` column shows the average number of cookies eaten.```{r boot-conf-cookies}# (Note that we have to use this function on the dataset of nested results, not# the one where we unnested everything.)cookie_bootstrap_stats %>%int_t(stats)```::: {.callout-tip}#### Distributions and percentiles`int_t()` technically works by using the critical values of the *t*-distribution at 2.5% and 97.5% (thus making a 95% confidence interval). If you want to be fancy and not assume that the distribution of the estimate follows a *t* distribution, you can use `int_pctl()` instead, which just finds the observed values at the 2.5% and 97.% percentiles without making any distributional assumptions.:::### Bootstrapping with inverse probability weightingPhew. Now that we get the rough intuition behind bootstrapping (create a bunch of random copies of the original data, calculate stuff on each copy, and collapse those results into a single number), we can go through the same process with inverse probability weighting to get standard errors and confidence intervals that incorporate the uncertainty of the propensity score model and are thus more accurate.We need to create a little function that will run the propensity score model, generate inverse probability weights, and run the outcome model for each of our random datasets. These are all the steps we ran earlier in the example, just in one function, running on a dataset named `current_data`:```{r one-ipw-function}fit_one_ipw <-function(split) {# Work with just a sampled subset of the full data current_data <-analysis(split)# Fit propensity score model model_net <-glm(net ~ income + temperature + health,data = current_data,family =binomial(link ="logit"))# Calculate inverse probability weights net_ipw <-augment_columns(model_net, current_data,type.predict ="response") %>%mutate(ipw = (net_num / .fitted) + ((1- net_num) / (1- .fitted)))# Fit outcome model with IPWs model_outcome <-lm(malaria_risk ~ net,data = net_ipw,weights = ipw)# Return a tidied version of the model resultsreturn(tidy(model_outcome))}```With that function, we can take our `nets` data and make 1000 random copies of it. We'll then apply our neat `fit_one_ipw()` function to each of the datasets```{r ipw-boot, cache=TRUE}ipw_bootstrap_stats <-bootstraps(nets, 1000, apparent =TRUE) %>%mutate(results =map(splits, fit_one_ipw))```If we look at the results, we'll see the version with all of the regression output nested inside little datasets inside cells:```{r show-ipw-boot-nested, cache=TRUE, message=FALSE}ipw_bootstrap_stats```If we unnest that column, we can see the actual results. Notice how there are 2,002 rows here instead of 1,001—each dataset has two values in it: the intercept and the coefficient for `netTRUE`.```{r show-ipw-boot-unnested, cache=TRUE, message=FALSE}ipw_bootstrap_stats %>%unnest(results)```Since we're interested in the effect of net usage, we'll filter the data so we only look at that coefficient:```{r show-ipw-boot-nested-net-only, cache=TRUE, message=FALSE}ipw_bootstrap_net <- ipw_bootstrap_stats %>%unnest(results) %>%filter(term =="netTRUE")ipw_bootstrap_net```Now we can use Rubin's Rules to collapse all these estimates into a single average and a single standard error:```{r ipw-net-mean-se}ipw_bootstrap_net %>%summarize(avg_net =mean(estimate),avg_se =sqrt(mean(std.error^2) +var(estimate)))``````{r extract-bootstrap-stats, include=FALSE}boot_stats <- ipw_bootstrap_net %>%summarize(avg_net =mean(estimate),avg_se =sqrt(mean(std.error^2) +var(estimate)))```The average net effect (or point estimate) of `r boot_stats$avg_net` is the same as what we found with the regular `lm()` model from before, but the standard error is now bigger (`r round(boot_stats$avg_se, 2)`) and more like the standard error we got with matching + weights (`r round(matching_results$net_true$std.error, 2)`).Finally we can find the confidence interval for the estimate with `int_t()`:```{r ipw-net-conf}ipw_bootstrap_stats %>%int_t(results) %>%filter(term =="netTRUE")``````{r extract-bootstrap-conf, include=FALSE}boot_conf <- ipw_bootstrap_stats %>%int_t(results) %>%filter(term =="netTRUE") %>%mutate(conf = glue::glue("[{round(.lower, 2)}, {round(.upper, 2)}]"))```*That's* the correct result. Using inverse probability weighting, using a mosquito net causes a `r boot_stats$avg_net` point reduction in malaria risk scores, with a standard error of `r round(boot_stats$avg_se, 2)` and a 95% confidence interval of `r boot_conf$conf`.